Критерии ароматичности органических соединений. Ароматичность, критерии ароматичности.Правило Хюккеля. Смотреть что такое «хюккеля правило» в других словарях
значениями длин одинарных и двойных связей. В молекуле бензола все длины связей равны (1.395 ангстрем), тогда как в сопряженных ациклических полиенах они чередуются. В бутадиене, например, длина связи C 1 -C 2 составляет 1,34 Å, а длина связи C 2 -C 3 - 1,48 Å . Как правило, длины связей имеют тенденцию к чередованию в неароматических соединениях, а не в ароматических структурах. Типичные значения длин наиболее важных связей ациклических соединений приведены в табл. Эти значения могут быть сопоставлены с величинами длин связей в некоторых гетероароматических системах, как показано на рис. 1.Таблица. Типичные длины одинарных и двойных связей (Å) между sp 2 -гибридизованными атомами
| C-C | 1.48 | C=C | 1.34 |
| C-N | 1.45 | C=N | 1.27 |
| C-O | 1.36 | C=O | 1.22 |
| C-S | 1.75 | C=S | 1.64 |
| N-N | 1.41 | N=N | 1.23 |
Длины связей в четырех шестичленных гетероциклах, показанных на рис. 1, будут промежуточными между значениями для одинарных и двойных связей. Длины связей С-С в трех шестичленных моноциклах различаются незначительно и близки к значениям для бензола. На этом основании мы можем сделать вывод, что в этих соединениях существует значительная циклическая делокализация π-электронов. Для пятичленных гетероциклов мы видим существенное чередование связей. Поскольку в молекулах содержатся различные гетероатомы, было бы неправомерно сравнивать длины связей, но можно сказать, что в кислородсодержащих гетероциклах в большей степени выражена локализация связей. Однако и в этих соединениях длины связей отличаются от длин связей «чистых» одинарных и двойных связей. Во всех этих пятичленных гетероциклических системах существует циклическая делокализация, но она меньше, чем для шестичленных гетероциклов. В двух представленных пятичленных бициклических системах (индоле и индолизи-не) степень локализации связей гораздо выше, чем, например, в пирроле, и эта закономерность наблюдается и в случае других конденсированных систем по сравнению с их моноциклическими аналогами.
Порядок связи можно иногда оценить с помощью вицинальных констант спин-спинового взаимодействия (КССВ) в спектрах ПМР. Например, КССВ Jab и Jbc на соседних атомах углерода a, b и с, удаленных от гетероатома, должны быть равны, если связи Сaа-Сb и Сb-Сc равны по длине. Значение отношения Jab:Jbc должно находиться в интервале от 0,5 до 1,0 в зависимости от степени чередования связей. Сравнивая эти величины в серии подобных соединений, можно определить значения степени локализации связей. Например, изменения в отношении Jab:Jbc для четырех соединений представленные на рис. 2, позволяют оценить степень фиксации связей в изоиндоле, для которого невозможно получить данные с помощью рентгеноструктурного анализа.
Эффекты кольцевых токов и химические сдвиги в спектрах ПМР
Химические сдвиги сигналов протонов в бензолах больше чем в аналогичных ациклических полиенах. В частности, это приписывают влиянию «диамагнитного кольцевого тока». Когда раствор бензоидного соединения помещают в магнитное поле, молекулы выстраиваются под правильными углами к полю, и возникает диамагнитный кольцевой ток из-за наличия делокализованных π-электронов. Это создает вторичное магнитное поле, которое противоположно приложенному полю внутри цикла, но усиливает его вне цикла (рис. 3). Таким образом, ядра водорода, лежащие в области выше или ниже центра кольца, экранированы, а лежащие на периферии - дезэкранированы. Изменения труднее наблюдать в спектрах 13 С-ЯМР, так как химические сдвиги 13 С гораздо больше, и дополнительное экранирование и дезэкранирова-ние, вызванное кольцевым током, относительно менее заметно.Существование диамагнитного кольцевого тока, проявляющееся в экранирующем и дезэкранирующем влиянии на химические сдвиги протонов, было предложено считать диагностическим тестом на ароматический характер соединения. Это оправдано тем, что установлена теоретическая связь между диамагнитной восприимчивостью и энергией резонанса. Но этот критерий следует использовать с осторожностью, так как эффекты кольцевого тока возрастают с увеличением размера цикла и, следовательно, довольно значительны в больших аннуленах и гетероаннуленах. С практической точки зрения, для того чтобы обнаружить экранирование и дезэкранирование, необходимо иметь для сравнения подходящие неароматические эталонные соединения, а такие соединения нелегко найти для некоторых гетероциклических систем. На химические сдвиги оказывают влияние некоторые другие факторы, помимо диамагнитного кольцевого тока, как, например, нарушение распределения π-электронов гетероатомом и влияние природы растворителей. Величины химических сдвигов для многих гетероциклов сильно зависят от природы растворителя. Однако мы можем видеть качественное влияние кольцевых токов, сравнивая спектры ПМР пиридина, фурана и тиофена и их дигидроаналогов (рис. 4).
Сравнения с неароматическими системами такого типа подвергаются критике, поскольку действительно трудно подобрать подходящие модельные соединения для некоторых простых гетероциклов, таких, как, например, пиррол. Используются непрямые методы оценки влияния диамагнитного кольцевого тока: например, величины химических сдвигов метильных групп гетероциклов, приведенные на рис. 5, сравнивали со значениями, рассчитанными для линейных моделей. Наблюдаемые сдвиги в слабое поле были приняты критерием оценки относительной ароматичности гетероциклов. Однако в основном «эффект кольцевого тока» следует рассматривать скорее как качественный индикатор ароматичности, чем количественный.
Другие физические методы изучения электронного строения
Существует несколько экспериментальных методов изучения электронных энергетических уровней или распределения электронной плотности. Они не могут считаться критериями ароматичности, но обеспечивают независимые экспериментальные оценки состоятельности расчетов энергий молекулярных орбиталей гетероциклов.Ультрафиолетовые спектры поглощения в течение многих лет используют как качественный метод для определения подобия характера связей в различных соединениях. Области, которые определяются π→π*-переходами, подобны таковым для карбоциклических аналогов, хотя спектры многих гетероциклов содержат дополнительные энергетические переходы, которые можно приписать n→π*-поглощению.
Энергетические уровни заполненных молекулярных орбиталей могут быть рассчитаны с помощью фотоэлектронной спектроскопии. Электроны выбрасываются с занятых молекулярных орбиталей при облучении молекул ультрафиолетовым светом высокой энергии в газовой фазе. Энергии этих электронов непосредственно связаны с потенциалами ионизации, обусловленными удалением электронов с различных молекулярных орбиталей. Анализ спектров включает определение спектральных областей электронных состояний молекулярных ионов и, следовательно, идентификацию орбиталей, с которых произошло испускание электронов. Таким образом, метод служит экспериментальным тестом для предсказанных изменений в уровнях связей серии гетероциклических соединений. Например, были получены аналоги пиридина, в которых атом азота был заменен другими элементами V группы (Р, As, Sb, Bi). Изучение фотоэлектронных спектров показало, что π-связывание в этих соединениях подобно π-связыванию в молекулах бензола и пиридина. π-Связывающие орбитали напоминают те, которые показаны на рис. 6. Энергии ионизации, связанные с уровнем π 2 (рис. 6), уменьшаются с увеличением размера гетероатома, так как он становится более электроположительным. Фотоэлектронный спектр силабензола также соответствует ожидаемому для бензольного аналога.
Дополнительный метод, дающий возможность измерить сродство к электрону и оценить энергетические уровни незанятых орбиталей, известен как спектроскопия электронного пропускания. Электрон из электронного пучка временно захватывается незанятой орбиталью молекулы, и образуется анион с очень коротким временем жизни (10 -12 -10 -15 с). Величины, характеризующие сродство к электрону, получают, анализируя изменение в спектре электронного рассеяния. Этим методом было определено сродство к электрону некоторых ароматических гетероциклов. С помощью этих методов были подтверждены данные, рассчитанные исходя из энергий π-орбиталей ароматических гетероциклов (представлены на рис. 6 и 7).
Термохимическая оценка ароматичности: Эмпирические энергии резонанса
Для оценки стабилизации ароматических соединений обычно используют два термохимических метода: измерение стандартной энтальпии сгорания и стандартной энтальпии гидрирования. Теплота сгорания пиридина, например, представляет собой изменение энтальпии в соответствии с уравнениемC 5 H 5 N (г.) + 25/4 O 2 → 5CO 2 (г.) + 5/2 H 2 O (ж.) + 1/2 N 2 (г.)
и значение может быть определено экспериментально с помощью калориметрии. Метод также может быть применен для определения экспериментального значения теплоты образования соединения. Атомная теплота образования пиридина представляет собой изменение энтальпии в соответствии с уравнением
C 5 H 5 N (г.) → 5С (г.) + 5Н (г.) + N (г.)
Величина может быть получена из теплоты сгорания, если использовать известные величины теплот сгорания и атомизации углерода, водорода и азота.
Теплота образования может быть рассчитана сложением индивидуальных значений энергий связей для молекулы: для пиридина это должны быть значения, соответствующие локализованной структуре (структуре Кекуле). Разница между экспериментальной (численно меньшей) и рассчитанной величинами и будет мерой стабилизации делокализованной системы; она называется эмпирической энергией резонанса. Полученные значения зависят от величин энергий связей, используемых в расчете, а также от выбора модельной системы «локализованной связи».
Теплоты гидрирования ароматических соединений можно использовать для расчета эмпирических энергий резонанса путем сравнения с экспериментальными значениями для подходящих модельных соединений. Например, сравним теплоту гидрирования бензола [ΔН = -49,7 ккал/моль (-208 кДж/моль)] с таковой для 3 молей циклогексена [ΔН = -28,4 ккал/моль (-119 кДж/моль), [ΔН = -85,3 ккал/моль (-357 кДж/моль)]. Разница 35,6 ккал/моль (149 кДж/моль) соответствует эмпирической энергии резонанса бензола. Немного иное значение получают, если модельную систему выбирают другим способом. Так, значения теплот гидрирования первой и второй двойных связей 1,3-циклогексадиена экстраполируют и получают величину теплоты гидрирования при добавлении третьего моля водорода к гипотетическому «циклогексатриену». Сумму трех значений затем принимают за величину для локализованной модели, как показано ниже (рассчитано экстраполяцией):
C 6 H 10 + H 2 → C 6 H 12 ΔН = -28,4 ккал/моль (-119 кДж/моль)
1,3-C 6 H 8 + H 2 → C 6 H 10 ΔH = -26,5 ккал/моль (-111 кДж/моль)
C 6 H 6 + H 2 → C 6 H 8 ΔН = -24,6 ккал/моль (-103 кДж/моль)
Суммарная теплота гидрирования этой локализованной модели составляет - 79,5 ккал/моль (- 333 кДж/моль), и, таким образом, эмпирическая энергия резонанса бензола равна 29,8 ккал/моль (125 кДж/моль).
Однако совершенно ясно, что не стоит придавать слишком большого значения абсолютный величинам энергий резонанса. Величины, полученные подобными методами для серии соединений, могут давать только приемлемую относительную оценку степени стабилизации. Большинство значений для гетероциклических соединений основано на величинах теплот сгорания, так как многие модельные системы, необходимые для измерения теплот гидрирования, трудно доступны. Литературные данные различаются в очень широком диапазоне, главным образом из-за выбранных значений энергий связей. Некоторые сравнимые значения (полученные подобными методами) представлены в табл.
Таблица. Эмпирические энергии резонанса
Молекулярные орбитали и энергия делокализации
Рассмотрим гетероциклы, полностью ненасыщенные и плоские или почти плоские, с замкнутым циклом атомов с взаимодействующими р-орбиталями. В приближении Хюккеля электроны на π-молекулярных орбиталях рассматриваются отдельно от электронов, расположенных на α-орбиталях. Энергии π-молекулярных орбиталей могут быть выражены с помощью двух констант. Первая, кулоновский интеграл, обозначаемый символом α, отражает приближенное значение силы притяжения электрона отдельного атома. В углеродной π-электронной системе α представляет собой энергию электрона на изолированной р-орбитали до перекрывания. Вторая константа, резонансный интеграл, означает меру стабилизации, достигаемую при взаимодействии соседних р-орбиталей. Эту величину обозначают символом β.Величины энергий шести π-орбиталей бензола, рассчитанные по методу Хюккеля, приведены на рис. 8, а. Две π-орбитали этилена представлены для сравнения на рис. 8, б. Шесть π-электронов, занимающих три связывающие орбитали бензола, имеют суммарную энергию (6α + 8β), тогда как шесть π-электронов на трех изолированных связывающих орбиталях этилена будут иметь общую энергию (6α + 6β). Таким образом, π-электронная система бензола более стабильна на величину 2β, которую называют энергией делокализации бензола. Очевидно, что энергия делокализации будет такой же и для пиридина, и для других шестичленных гетероциклов, если игнорировать эффект, возникающий при замене атома углерода на атом азота. На практике такие эффекты могут быть компенсированы использованием параметров, вносящих поправку на неравномерное распределение π-электронной плотности.
Эта энергия делокализации не соответствует эмпирической энергии резонанса, так как последняя рассчитывается для модели с чередующимися длинами связей, а первая основывается на гипотетической модели локализации с геометрией, идентичной геометрии де-локализованной системы. Для того чтобы установить соотношение между ними, мы должны к эмпирической энергии резонанса добавить энергию, необходимую для сжатия структуры с чередующимися простыми и кратными связями до структуры с нечередующимися связями. Эта энергия деформации, рассчитанная для бензола, составляет 27 ккал/моль (113 кДж/моль), т. е. весьма существенную величину по сравнению с эмпирической энергией резонанса. Следовательно, полезнее признать, что энергии делокализации представляют собой относительные величины, чем определять их численные значения.
Рассчитанные энергии резонанса
Проблема измерения ароматической стабилизации на основании модели простой несопряженной π-электронной системы состоит в том, что «энергия делокализации» не является уникальным свойством циклических систем. Например, на основе простого метода МО Хюккеля можно показать, что энергия делокализации бутадиена составляет 0,472β; другие ациклические сопряженные системы также имеют некоторую энергию делокализации. Пытаясь найти меру ароматичности, необходимо оценивать дополнительный вклад в общую энергию делокализации вследствие того, что соединение имеет циклическую структуру. В связи с этим было высказано предположение, что при расчете энергии резонанса следует использовать энергии связей неароматических систем, а не несопряженных систем в качестве эталонных структур. Было показано, что энергия π-связи линейных полиенов прямо пропорциональна длине цепи. Каждая дополнительная «простая» или «двойная» связь С-С в полиене вносит в общую π-энергию такой же вклад, как и в случае бутадиена или гексатриена. Это, конечно, не означает, что отсутствует сопряжение, но показывает, что сопряжение также влияет на энергию связи в нециклических системах. Следовательно, можно рассчитать «эталонные» энергии π-связей для любой циклической или ациклической π-системы, складывая величины, соответствующие определенным типам связей. Этот аддитивный принцип применим к π-связям с гетероатомами в такой же степени, как и к связям углерод-углерод.Циклические системы, в которых наблюдается дополнительная энергия π-связи по сравнению с рассчитанными эталонными величинами, называют «ароматическими». Дополнительная энергия стабилизации была названа «резонансной энергией Дьюара», но принцип расчета энергий резонанса был принят позднее. Циклические системы, энергии резонанса которых близки к нулю [не более 2,5 ккакл/ моль (10 кДж/моль)], относят к «неароматическим». Несколько циклических систем, для которых рассчитанная энергия резонанса имеет отрицательную величину (они обладают меньшей энергией связи, чем эталонная структура), называют «антиароматическими».
Энергии резонанса, основанные на модели Дьюара, можно рассчитать методом МО Хюккеля, несмотря на то, что метод игнорирует σ- и π-взаимодействия. Это обусловлено тем, что σ- и π-вклады в энергию связи прямо пропорциональны порядку данной связи. Следовательно, π-резонансные энергии прямо пропорциональны общим энергиям резонанса. Что касается связей с гетероатомом, то величины кулоновского и резонансного интегралов необходимо модифицировать. При этом должны быть получены величины, наилучшим образом совпадающие с экспериментальными значениями теплот атомизации известных соединений, которые затем используют для расчета энергий различных типов π-связей в единицах измерения резонансного интеграла β. Общую энергию π-связи эталонной структуры (т. е. структуры с преобладающей валентностью) рассчитывают сложением вкладов индивидуальных связей, которые затем сравнивают с общей энергией т-связи, вычисленной по методу МО Хюккеля.
Для того чтобы провести сравнение ароматичности других гетероциклов, удобно рассчитывать энергию резонанса на один π-электрон (РЭЭ) путем деления энергии резонанса на число π-электронов в молекуле. Для известных систем эти значения хорошо коррелируют с другими критериями ароматичности; некоторые данные для важнейших гетерциклов приведены в табл. Метод можно также использовать для предсказания степени ароматичности еще не синтезированных гетероциклических соединений. В табл. также приведены некоторые рассчитанные значения энергий ароматизации, которые представляют собой разницу энергий аналогов с локализованными и делокализованными структурами.
Таблица. Резонансные энергии в расчете на одни π-электрон (РЭЭ) и энергии ароматизации "некоторых гетероциклических соединений"
Ароматичность - понятие, характеризующее совокупность особых структурных, энергетических и магнитных свойств, а также особенностей реакционной способности циклических структур с системой сопряженных связей.
Хотя ароматичность - одна из важнейших и наиболее плодотворных концепций химии (не только органической), - не существует общепринятого краткого определения этого понятия. Ароматичность понимается через совокупность особых признаков (критериев), присущих ряду циклических сопряженных молекул в той или иной мере. Часть этих критериев имеет экспериментальную, наблюдаемую природу, но другая часть основывается на квантовой теории строения молекул. Ароматичность имеет квантовую природу.
Невозможно объяснить ароматичность с позиций классической структурной теории и теории резонанса.
Не следует путать ароматичность с делокализацией и сопряжением.
В молекулах полиенов (1,3-бутадиена, 1,3,5-гексатриена и т.п.) проявляется явно выраженная тенденция к делокализации электронов и образованию единой сопряженной электронной структуры, что проявляется в спектрах (в первую очередь, электронных спектрах поглощения), некотором изменении длин и порядков связей, энергетической стабилизации, особых химических свойствах (электрофильное 1,4-присоединение в случае диенов и пр.). Делокализация и сопряжение - необходимые, но не достаточные условия ароматичности. Можно дать определение ароматичности как свойства, при котором сопряженное кольцо ненасыщенных связей проявляет бόльшую стабильность, чем ту, которую можно было бы ожидать только при одном сопряжении. Однако этим определением нельзя пользоваться, не имея экспериментальных или расчётных данных по стабильности циклической сопряжённой молекулы.
Для того чтобы молекула могла быть ароматической, она должна содержать хотя бы один цикл, каждый из атомов которого располагает пригодной для образования ароматической системы р-орбиталью. Ароматическим в полном смысле этого слова считается (в случае выполнения критериев, перечисленных ниже) именно этот цикл (кольцо, система колец).
В этом цикле должно быть 4n+2
(то есть 2, 6, 10, 14, 18, 22 и т.п.) p-электронов
.
Это правило называется правилом или критерием ароматичности Хюккеля
. Источник этого правила - сильно упрощенные квантовохимические расчеты идеализированных циклических полиенов, произведенные на заре развития квантовой химии. Дальнейшие исследования показали, что в основе своей это простое правило дает верные предсказания ароматичности даже и для очень сложных реальных систем.
Правилом, тем не менее, нужно правильно пользоваться, иначе прогноз может быть неверен.
Какие орбитали считаются пригодными для образования ароматической системы? - Любые орбитали, перпендикулярные плоскости цикла, и
а) принадлежащие входящим в цикл кратным (эндоциклическим двойным или тройным) связям;
б) соответствующие неподеленным парам электронов у гетероатомов (азота, кислорода, и т.п.) или карбанионов;
в) соответствующие шестиэлектронным (секстетным) центрам, в частности карбокатионам.
Критерии ароматичности.
Энергетический
(повышение термодинамической устойчивости за счет делокализации электронов, так называемая энергия делокализации - ЭД).
Можно представить бензол производным трёх молекул этилена и сравнить энергии исходных фрагментов и конечной молекулы. У каждой молекулы этилена по 2 p-электрона (всего 6) на молекулярных орбиталях (МО) одинаковой энергии (α+β), а у бензола 6 электронов располагаются на трёх связывающих молекулярных орбиталях, давая в сумме более отрицательное значение энергии системы (α и β меньше 0).
Очевидное энергетическое преимущество составляет 2β = 36 ккал/моль или 1,56 эВ - это ЭЭР (эмпирическая энергия резонанса).
Энергетический критерий из всех самый неудобный и неясный. Величины энергий для этого критерия берут всегда расчетные, потому что, как правило, невозможно подобрать соответствующую неароматическую молекулу для сравнения. Следует, поэтому, спокойно относиться к тому, что существует множество различных оценок энергии делокализации даже для классических ароматических молекул, а для более сложных систем эти величины вообще отсутствуют. Никогда нельзя сравнивать разные ароматические системы по величине энергий делокализации - нельзя сделать вывод, что молекула А ароматичнее молекулы В, потому что энергия делокализации больше.
Структурный
- очень важный, если не самый важный, критерий, так как имеет не теоретическую, а экспериментальную природу. Специфика геометрии молекул ароматических соединений заключается в тенденции к копланарному расположению атомов и выравниванию длин связей. У бензола выравнивание длин связей идеально - все шесть С-С связей одинаковы по длине. У более сложных молекул выравнивание не идеально, но значительно. В качестве критерия берут меру относительного отклонения длин сопряженных связей от среднего значения. Чем ближе к нулю, тем лучше. Эту величину можно проанализировать всегда, если имеется структурная информация (экспериментальная или из высококачественного квантовохимического расчета). Тенденция к копланарности обуславливается выгодностью параллельного расположения осей атомных р-орбиталей для их эффективного перекрывания.
Магнитный
(наличие кольцевого тока - диатропная система, влияние на химические сдвиги протонов снаружи и внутри кольца, примеры - бензол и -аннулен). Самый удобный и доступный критерий, так как для его оценки достаточно спектра 1H ЯМР. Для точного определения используют теоретические расчеты химических сдвигов.
Химический
- склонность к реакциям замещения, а не присоединения. Самый наглядный критерий, ясно различающий химию ароматических соединений от химии полиенов. Но работает он далеко не всегда. В ионных системах (например, в циклопентадиенил-анионе или тропилий-катионе) замещение наблюдать невозможно. Реакции замещения иногда проходят и на неароматических системах, а ароматические всегда в какой-то степени способны к реакциям присоединения. Поэтому химический критерий более правильно назвать признаком ароматичности.
Представление энергии ароматической системы.
Общая формула:
E j (энергия орбитали j уровня) = α + m j β
α - кулоновский интеграл, энергия С2р орбитали,
β - резонансный интеграл, энергия взаимодействия 2-х атомных орбиталей на соседних атомах
m j = 2сos(2jπ/N), где N- число атомов углерода в цикле.
Наиболее простым и наглядным графическим изображением энергии являтся круг Фроста . Для его построения необходимо вписать в окружность ароматическую молекулу, направив вершиной вниз, тогда точки соприкосновения многоугольника и окружности будут соответствовать энергетическим уровням МО. По вертикали наносится энергетическая шкала, все уровни ниже горизонтального диаметра - связывающие, выше - разрыхляющие. Электроны заполняют от самой нижней орбитали согласно правилу Паули.

Наиболее выгодным будет такое состояние, когда полностью заполнены все связывающие орбитали.
Позднее появилось ещё множество предположений о структуре бензола:

Однако даже до сих пор молекула C 6 H 6 продолжает преподносить сюрпризы. Бодриков И.В.: «Я вынужден констатировать, что сейчас в мире нет человека, кто бы знал, что такое бензол» (2009)

(один из водородов перемещается в положение, перпендикулярное кольцу)
Понятия «ароматичность» и «антиароматичность» в химии не имеют отношения к наличию запаха у вещества. Ароматические соединения - это вещества, в которых замкнутая сопряженная система из π-электронов, образующаяся в результате перекрывания р-электронных облаков соседних атомов, придает молекуле аномально высокую стабильность. Самое, пожалуй, известное ароматическое соединение - бензол С 6 Н 6 , гораздо более инертный по сравнению с соединениями, в которых на углерод приходится большее число атомов водорода - С 6 Н 8 , С 6 Н 10 , С 6 Н 12 и С 6 Н 14 . В антиароматических соединениях замкнутая сопряженная система из π-электронов, напротив, делает антиароматическую молекулу крайне неустойчивой, а в ряде случаев антиароматические соединения с желаемой формулой вообще невозможно получить.
На основании свойств бензола и представлениях о том, что углерод образует четыре химические связи, а также о том, что атомы углерода способны связываться друг с другом двойными и тройными связями, было предложено несколько ошибочных вариантов структурных формул для бензола. Уже в ХХ веке все соединения, соответствующие этим формулам, были синтезированы. Было установлено, что их физические и химические свойства отличаются от свойств бензола. Также было обнаружено, что призман (бензол Ладенбурга) и бензол Дьюара самопроизвольно превращаются в бензол при комнатной температуре.

Правильную - циклическую - формулу этого соединения удалось установить Фридриху Августу Кекуле , который, судя по его воспоминаниям, пришел к этой идее во сне: ему приснилась змея, кусающая себя за хвост, и он решил, что бензол должен представлять собой шестичленный цикл из атомов углерода с чередующимися двойными и одинарными связями.
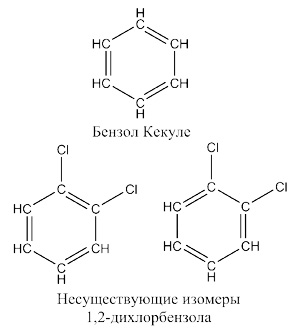
Правда, в соответствии с формулой Кекуле - и он сам так предполагал, - должно было бы существовать два изомера 1,2-дихлорбензола - в одном атомы хлора должны были быть разделены двойной связью, в другом - одинарной. Поскольку, как оказалось позже, в бензоле нет чередующихся двойных и одинарных связей, все атомы углерода эквивалентны, а π-электроны образуют единую замкнутую систему, такие изомеры и не могут существовать. Естественно, что их так и не удалось обнаружить, а сам Кекуле предполагал, что неудачи обнаружения этих изомеров связаны с очень быстрым превращению их друг в друга.
Именно Кекуле и ввел понятие «ароматичность», которое предложил рассматривать, как особую характеристику бензола и родственных по структуре соединений - их устойчивость и склонность участвовать в реакциях замещения, а не присоединения. Такое химическое поведение нельзя было объяснить формулой Кекуле, предполагавшей наличие в бензольном цикле трех двойных связей - в других соединениях, не относившихся к ароматическому ряду, кратные связи активно вступали в реакции присоединения.
В чем состоит причина такого поведения ароматических соединений удалось объяснить в 1931 году Эриху Хюккелю , который, использовав для изучения бензола упрощенный метод квантовой механики - метод молекулярных орбиталей , показал, что в молекуле бензола нет двойных и одинарных связей углерод–углерод, а р -электроны атомов углерода образуют единое кольцевое электронное облако, которому Хюккель дал название «ароматическая система». Он же разработал и фигурирующие в условии задачи правила, позволяющие относить молекулы к ароматическим или антиароматическим. Два правила Хюккеля одинаковы и для ароматических и для антиароматических соединений: плоское строение молекулы и замкнутая сопряженная система нужны для образования кольцевого электронного облака, а разное количество электронов в таком облаке может стабилизировать или дестабилизировать молекулу.
Хотя ароматичность - одна из важнейших концепций химии, пока нет общепринятого краткого определения этого понятия. Ароматичность понимается как совокупный набор особых признаков, в той или иной мере присущих ряду циклических сопряженных молекул. Часть этих признаков можно наблюдать экспериментально, но другие признаки можно описать, только основываясь на квантовой теории строения молекул (сюда как раз относится правило Хюккеля о числе электронов в замкнутой системе). Невозможно объяснить ароматичность с позиций только классической структурной теории химии.
Экспериментально наблюдать можно следующие проявления ароматичности:
1) Химическое - склонность ароматической молекулы к реакциям замещения , а не присоединения .
2) Структурный - тенденция к плоскому расположению атомов и выравниванию длин связей . У бензола выравнивание длин связей идеально - все шесть связей углерод-углерод одинаковы. У более сложных молекул выравнивание длин связей не идеально, но значительно.
3) Магнитный - замкнутая электронная система ароматических соединений оказывает влияние на параметры спектров ядерного магнитного резонанса ароматических соединений. В ароматических соединениях при приложении к ним внешнего магнитного поля возникает кольцевой ток, который способствует возникновению внутри молекул локального магнитного поля, направленного противоположно внешнему. В результате взаимодействия внутреннего и внешних магнитных полей экранирование атомов водорода и атомов углерода ароматической системы понижается, и их сигналы регистрируются в области слабых полей, которая обычно и называется «областью сигналов ароматических атомов водорода» для ЯМР-спектроскопии 1 Н и «областью сигналов ароматических атомов углерода» для ЯМР-спектроскопии 13 С.
Кроме бензольного кольца и углеводородов ароматические свойства проявляют многие гетероциклические соединения - пиррол , фуран , тиофен , пиридин , индол , оксазол и другие. При этом в сопряженную систему шестичленных гетероциклов гетероатом отдает один электрон (по аналогии с углеродом), в 5-атомных - неподеленную электронную пару. Ароматические свойства могут проявлять не только нейтральные молекулы, но и заряженные частицы, например, циклопропенилий-катион (2π-электронная система), циклопентадиенил-анион (6π-электронная система), циклогептатриенил-катион (6π-электронная система), циклооктатетраенил-дианион (10π-электронная система).

В ряде случаев ароматические соединения получают из неароматических веществ в ходе процесса ароматизации. Наибольшее практическое значение имеет каталитический риформинг бензиновых фракций, который увеличивает октановое число моторного топлива (см. послесловие к задаче
Ароматичность
Ароматичность - особое свойство некоторых химических соединений, благодаря которому сопряженное кольцо ненасыщенных связей проявляет аномально высокую стабильность; большую чем та, которую можно было бы ожидать только при одном сопряжении .
Ароматичность не имеет непосредственного отношения к запаху органических соединений, и является понятием, характеризующим совокупность структурных и энергетических свойств некоторых циклических молекул, содержащих систему сопряженных двойных связей. Термин «ароматичность» был предложен потому, что первые представители этого класса веществ обладали приятным запахом.
К ароматическим соединениям относят обширную группу молекул и ионов разнообразного строения, которые соответствуют .
История
Критерии ароматичности
Существуют несколько критериев, по которым молекула может быть отнесена к ароматическим.
Правило Хюккеля
Ароматическими являются молекулы, подчиняющиеся правилу Хюккеля : ароматической является плоская моноциклическая сопряженная система, содержащая (4n + 2)π-электронов (где n = 0,1,2…). Это правило выводится непосредственно из квантово-химических вычислений МОХ.
Современные представления
В современной физической органической химии выработано общая формулировка критерия ароматичности .
| Ненасыщенная циклическая или полициклическая диатропная молекула или ион может рассматриваться как ароматическая, если все атомы цикла входят в полностью сопряженную систему таким образом, что в основном состоянии все π-электроны располагаются только на связывающих молекулярных орбиталях аннулярной (замкнутой) оболочки. |
Ароматические соединения
Ароматизация
Ароматизация - образование ароматических соединений из соединений других типов.
В промышленности широко применяют процессы ароматизации продуктов переработки нефти для увеличения содержания в них ароматических углеводородов. Наибольшее значение имеет каталитический риформинг бензиновых фракций.
Процессы ароматизации протекают в условиях биохимического синтеза в растениях, животных, грибах и микроорганизмах. Одним из наиболее существенных метаболических путей, неотъемлемой частью которого выступают реакции ароматизации, является шикиматный путь .
Источники
- Реутов О.А. Органическая химия. - М .: Изд-во МГУ, 1999. - Т. 2. - 624 с. - ISBN 5-211-03491-0
- Агрономов А.Е. Избранные главы органической химии. - 2-е. - Москва: Химия, 1990. - 560 с. - ISBN 5-7245-0387-5
- Горелик М.В. Современное состояние проблемы ароматичности // Успехи химии . - 1990. - Т. 59. - № 2. - С. 197-228.
Примечания
| Химическая связь | |||||||
|---|---|---|---|---|---|---|---|
|
|||||||
| Межмолекулярное взаимодействие |
| ||||||
В органической химии хорошо известно и широко используется такое понятие, как ароматичность некоторых органических соединений. Термин «ароматичность» связан прежде всего с бензолом, его гомологами и многочисленными производными. Этот термин относится исключительно к структуре молекул этих веществ, их свойствам, но не имеет никакого отношения к их запаху. Правда, первые ароматические соединения имели, вероятно, приятный запах (некоторые натуральные эфиры, душистые смолы, например ладан и др.).
Ароматичность - общий признак некоторых циклических органических соединений, обладающих совокупностью особых свойств.
Наличие единой замкнутой системы π-электронов в молекуле - основной признак ароматичности.
Ароматические соединения подчиняются правилу Э. Хюккеля (1931):
Плоские моноциклические соединения, имеющие сопряженную систему π-электронов, могут быть ароматическими, если число этих электронов равно 4 n +2 (где n = 0,1,2,3, 4 и т.д., т.е. число π-электронов в молекуле может быть 2, 6, 10, 14, 18 и т.д.).
Эти особенности обусловливают все важнейшие физические и химические свойства ароматических соединений. Например, они вступают преимущественно в реакции замещения (в основном электрофильного), а не присоединения (несмотря на формальную ненасыщенность). Ароматические соединения обладают высокой устойчивостью, например к окислителям. Их молекулы имеют плоское строение. Если же это требование не выполняется, то в молекуле нарушается параллельность осей 2р-орбиталей, что приводит к устранению сопряжения и, как следствие, к нарушению выравненности π-электронной плотности в системе.
Номенклатура
Систематическое название всех ароматических углеводородов - арены , а бензола - бензен . Гомологи бензола рассматривают как замещенные бензола и цифрами указывают положение заместителей. Однако систематическая номенклатура допускает название «бензол», а для некоторых гомологов бензола - тривиальные названия: винилбензол (I) называют стиролом , метилбензол (II) - толуолом, диметилбензол (III) - ксилолом, изопропилбензол (IV) - кумолом, метоксибензол (V) - анизолом и т.д.:
Ароматические радикалы имеют общее название - арилы (Аr). Радикал С 6 Н 5 - называют фенилом (от старого названия бензола - «фен»).
Изомерия.
Общая формула гомологов бензола С n Н 2 n -6 . Все шесть атомов водорода в молекуле бензола одинаковы и при замещении одного из них на один и тот же радикал образуется одно и то же соединение. Поэтому однозамещенный бензол изомеров не имеет. Например, существует только один метилбензол:

При замещении двух атомов водорода на метальные группы образуются три изомера - ксилолы , которые отличаются друг от друга расположением заместителей в кольце:



орто -диметилбензол, мета -диметилбензол, пара -диметилбензол,
или 1,2-диметилбензол или 1,3-диметилбензол или 1,4-диметилбензол
(о -ксилол) (м -ксилол) (п -ксилол)
Вместо буквенного обозначения (орто-, мета-, пара -, или сокращенно: о-,м-, п-) можно пользоваться цифровым: 1,2-, 1,3-, 1,4-. Изомеры могут отличаться характером заместителей:


пропилбензол изопропилбензол